
Σύγχρονες τάσεις στη θεραπεία της μεσογειακής αναιμίας (θαλασσαιμίας): Η επιλογή της γονιδιακής θεραπείας
1. Οι αιμοσφαιρινοπάθειες
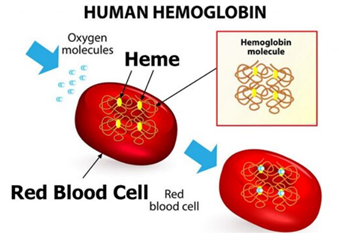
Η αιμοσφαιρίνη είναι η κύρια πρωτεΐνη στα ερυθρά αιμοσφαίρια και επιτελεί τον ρόλο της μεταφοράς του οξυγόνου. Η αιμοσφαιρίνη αποτελείται από τέσσερις πολυπεπτιδικές αλυσίδες, δύο α-σφαιρίνης, και δύο β-σφαιρίνης, και από τέσσερα μόρια αίμης (προσθετική ομάδα σιδήρου) (εικόνα 1). Η μετάλλαξη του γονιδίου της αιμοσφαιρίνης προκαλεί ασθένειες οι οποίες κληρονομούνται από τους γονείς στους απογόνους, και επειδή αφορούν την αιμοσφαιρίνη ονομάζονται αιμοσφαιρινοπάθειες (hemoglobinopathies). Οι ασθένειες αυτές διακρίνονται στις: α-θαλασσαιμία, β-θαλασσαιμία και δρεπανοκυτταρική αναιμία (Sickle Cell Disease, SCD) (Elisabeth Kohne, 2016).
Η πιο κοινή από τις αιμοσφαιρινοπάθειες είναι η θαλασσαιμία ή μεσογειακή αναιμία, με τη σοβαρότερη επίπτωση αυτής να είναι η αναιμία, λόγω της μειωμένης ποσότητας ερυθρών αιμοσφαιρίων. Υπολογίζεται ότι κάθε χρόνο 1 στα 100.000 άτομα, σε όλο τον κόσμο, θα πάσχουν από την ασθένεια και ότι στην Ευρώπη θα πάσχει 1 στα 10.000 άτομα (Renzo G. and Raffaella O., 2010). Οι θαλασσαιμίες διακρίνονται σε α-θαλασσαιμίες και β-θαλασσαιμίες, ανάλογα με το αν το γενετικό ελάττωμα ή η διαγραφή αφορά το γονίδιο της α- ή της β-σφαιρίνης. Έτσι, οι ασθενείς με α-θαλασσαιμία έχουν δομικά φυσιολογική αλυσίδα α-σφαιρίνης, αλλά η παραγωγή της είναι μειωμένη. Ομοίως, οι ασθενείς με β-θαλασσαιμία έχουν δομικά φυσιολογική αλυσίδα β-σφαιρίνης, αλλά η παραγωγή της είναι μειωμένη. Καθένας από τους δύο κύριους τύπους θαλασσαιμίας μπορεί να παρουσιαστεί ως ετερόζυγη (ήπια) ή ομόζυγη (σοβαρή) κατάσταση. Η πρώτη είναι γενικά ασυμπτωματική, ενώ η δεύτερη είναι σοβαρή συγγενής αιμολυτική αναιμία.
Ο τρόπος ζωής των ασθενών που έχουν ενδιάμεση ή δριμύ (major) θαλασσαιμία είναι δύσκολος λόγω των συνεχών μεταγγίσεων αίματος. Η επίπτωση των μεταγγίσεων είναι η συγκέντρωση μεγάλων ποσοτήτων αίματος και σιδήρου στα όργανα και κυρίως στην καρδία δημιουργώντας καρδιακή ανεπάρκεια από νεαρή ηλικία. Ουσιώδεις επιπτώσεις στην υγεία των πασχόντων είναι μεταξύ άλλων ο υποθυρεοειδισμός, πρόκληση θρομβώσεων, σπληνομεγαλία, κ.α. (Εικόνα 2).
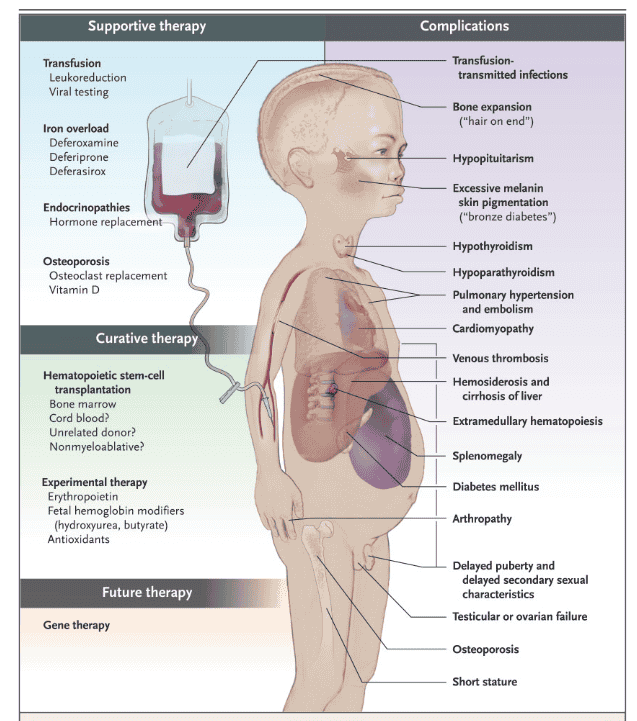
Μία κύρια θεραπεία των ασθενειών αυτών είναι η χορήγηση παραγόντων (όπως η δεσφεροξαμίνη και η δεφεριπρόνη) που δεσμεύουν τα υψηλά επίπεδα σιδήρου εξαιτίας των συχνών μεταγγίσεων. Σε κλινικές θεραπείες βρίσκεται το φάρμακο ΄΄Luspatercept΄΄, αναστολέας της σηματοδότησης TGF-β, οδηγώντας σε ενίσχυση της ερυθροποίησης (παραγωγή ερυθρών αιμοσφαιρίων στον οργανισμό) (Maria D. C. et al., 2018). Η μεταμόσχευση αιμοποιητικών βλαστοκυττάρων (πρόδρομων κυττάρων του οργανισμού ικανών να παράγουν διάφορα είδη κυττάρων) είναι η πιο αποτελεσματική μέθοδος, αν και η εύρεση συμβατών δοτών είναι επίπονη. Μία εναλλακτική προσέγγιση είναι η χρήση παραγόντων (όπως το Benserazide) για επαγωγή της έκφρασης εμβρυικών σφαιρινών (όπως η γ-σφαιρίνη) για να αντικαταστήσουν το ελλαττωματικό γονίδιο της β-σφαιρίνης (Arielle L. L. and Erica B. E., 2021).
Ωστόσο, η θεραπευτική αντιμετώπιση της θαλασσαιμία είναι υπό συνεχή μελέτη, τόσο προ-κλινικά όσο και κλινικά, με στόχο να βρεθεί μια πλήρως αποτελεσματική θεραπεία.
2. Η Γονιδιακή θεραπεία
Τα τελευταία χρόνια εξαιτίας της ραγδαίας προόδου στη μοριακή βιολογία και βιοϊατρική, γίνεται μεγάλη προσπάθεια για την χρήση της γονιδιακής θεραπείας στη θεραπευτική των θαλασσαιμιών. Η γονιδιακή θεραπεία έγκειται στη χρήση γενετικού υλικού για την θεραπεία ή την πρόληψη ασθενειών. Τα γονίδια είναι τμήματα του DNA που κωδικοποιούν μια λειτουργική πρωτεΐνη, απαραίτητη για τον οργανισμό. Η γονιδιακή θεραπεία επεμβαίνει είτε για να διορθώσει ένα γονίδιο, είτε να προσθέσει μέρη που μπορεί να λείπουν ή ακόμη και να διαγράψει γονίδιο, ή αποσιωπήσει την έκφραση μίας πρωτεΐνης (Εικόνα 3).
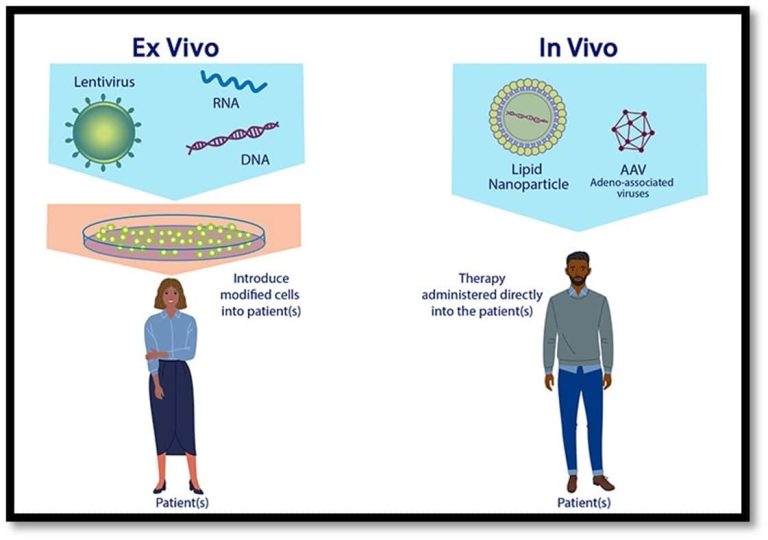
Η γονιδιακή θεραπεία μπορεί να γίνει άμεσα στον οργανισμό (in vivo) αλλά και έμμεσα (ex vivo) μέσω τροποποίησης των κυττάρων, εκτός του οργανισμού. Και στις δύο περιπτώσεις χρησιμοποιούνται φορείς (vectors), που μπορεί να νανοσωματίδια, ιοί ή μέσα μεταγωγής του γενετικού υλικού (Sofia K. Georgiou-Siafis et al., 2022).
Οι ιϊκοί φορείς πρέπει να είναι αδρανοποιημένοι ώστε να μην είναι παθογόνοι και μπορεί να είναι DNA ιοί, με απευθείας ενσωμάτωση στο γενετικό υλικό των ανθρώπινων κυττάρων αλλά και RNA ιοί, για ενσωμάτωση αφού προηγηθεί η αντίστροφη μεταγραφή του γενετικού υλικού. Η in vivo γονιδιακή θεραπεία πραγματοποιείται με φορείς οι οποίοι περιέχουν το σωστό γονίδιο και χορηγούνται απευθείας στον άνθρωπο με ένεση. Από την άλλη πλευρά, η ex vivo γονιδιακή θεραπεία χρειάζεται αρχικά να αφαιρεθούν τα κύτταρα από τον οργανισμό, έπειτα να τροποποιηθούν εκτός του οργανισμού και μετά, τα γενετικά διορθωμένα-τροποποιημένα κύτταρα να επιστρέψουν ξανά στον οργανισμό (American Society of Gene + Cell Therapy (ASGCT), 2023). (Εικόνα 4)
Παρόλα αυτά, και στις δύο περιπτώσεις υπάρχουν μειονεκτήματα και κίνδυνοι που μπορεί να παρουσιαστούν. Στην in vivo γονιδιακή θεραπεία υπάρχει ο κίνδυνος να γίνεται σύνθεση πρωτεϊνών του ιού και να προκαλέσει νόσο ή ακόμη και να δημιουργηθεί ένας ανασυνδυασμένος ιός, εάν δεν έχει αδρανοποιηθεί σωστά. Επίσης μπορεί να υπάρξει υπέρ-έκφραση μίας πρωτεΐνης με συνέπεια την τοξικότητα. Ο κίνδυνος ενσωμάτωσης του γονιδίου σε λάθος θέση στο γονιδίωμα του ασθενούς, μπορεί να οδηγήσει ακόμη και σε αναδιάταξη του χρωμοσώματος-στόχου, με πιθανά προβλήματα ακόμη και την καρκινογένεση. Στην ex vivo γονιδιακή θεραπεία έχουμε το μειονέκτημα ότι δεν μπορούμε να στοχεύσουμε εσωτερικά όργανα του οργανισμού. Γενικότερα, όμως υπάρχουν δυσκολίες στην γονιδιακή θεραπεία όπως είναι το κόστος αλλά και ελλοχεύουν κίνδυνοι για πιθανές βραχυπρόθεσμες ή μακροπρόθεσμες συνέπειες, όπως η ανοσοαπόκριση, καθώς η γονιδιακή θεραπεία βρίσκεται ακόμη υπό διερεύνηση.
3. Η γονιδιακή θεραπεία στην β-θαλασσαιμία
Zynteglo: betibeglogene autotemcel ή beti-cel
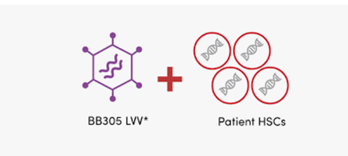
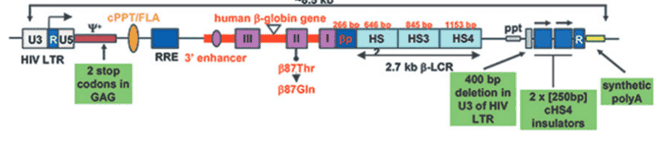
Στις 17 Αυγούστου 2022, ένα μεγάλο επίτευγμα στον επιστημονικό κόσμο ήταν η έγκριση μιας εφάπαξ γονιδιακής θεραπείας με βάση τροποποιημένα κύτταρα (Zynteglo: betibeglogene autotemcel ή beti-cel) από τον Οργανισμό Τροφίμων και Φαρμάκων (FDA). Το Betibeglogene autotemcel αξιολογείται ως γονιδιακή θεραπεία σε ασθενείς με β-θαλασσαιμία, που εξαρτώνται από τη μετάγγιση (Asghar AA, et al, 2022). Είναι μια πιθανή θεραπευτική θεραπεία για τη διόρθωση της ανισορροπίας της αλυσίδας β-σφαιρίνης, βελτιώνοντας έτσι την παραγωγή φυσιολογικής αιμοσφαιρίνης, την ερυθροποίηση και τη χρόνια αναιμία (Zakaria NA, et al, 2022). Το Beti-cel προσθέτει λειτουργικά αντίγραφα ενός τροποποιημένου γονιδίου HBB (αιμοσφαιρίνης β-hemoglobin B) που έχει υποκατάσταση αμινοξέος (T→Q) στη θέση 87, και προσθέτει επίσης ρυθμιστικά στοιχεία β-σφαιρίνης, επί αιμοποιητικών βλαστοκυττάρων (CD34 +κύτταρα). Η μεταγωγή του γονιδίου γίνεται μέσω απενεργοποιούμενο φακοϊικού φορέα (BB305) (Σχήμα 5).
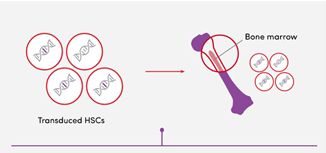
Αρχικά, λαμβάνει χώρα συλλογή αιμοποιητικών βλαστοκυττάρων από τους ασθενείς με στόχο τα βλαστοκύτταρα να τροποποιηθούν ex vivo και να εισαχθούν πίσω στον ασθενή (αυτόλογη μεταμόσχευση). Τα συλλεγμένα κύτταρα μετατρέπονται ex vivo με αυτο-απενεργοποιούμενους φακοϊικούς φορείς, οι οποίοι εισάγουν ένα ‘’construct’’ που έχει γονίδιο σφαιρίνης και άλλα στοιχεία που απαιτούνται για την έκφραση. Στη συνέχεια, αυτά τα κύτταρα εισάγονται στον ασθενή (που έχει υποστεί χημική καταστροφή του μυελού των οστών), όπου αναπαράγονται, με στόχο να δημιουργεί ένας νέος πληθυσμός των αιμοποιητικών κυττάρων (Nualkaew T, et al, 2021).
Μια πρόσφατη κλινική μελέτη το 2022, αξιολόγησε την αποτελεσματικότητα και την ασφάλεια του beti-cel σε 23 ασθενείς με μεταγγιζο-εξαρτώμενη β-θαλασσαιμία, από τους οποίους 20 έδειξαν ανεξαρτησία από μετάγγιση, μετά το πέρας της γονιδιακής θεραπείας. Συγκεκριμένα, τα μέσα επίπεδα αιμοσφαιρίνης προσδιορίστηκαν στα 11,7 g/dl, μετά από 12 μήνες. Οι πιο συχνές παρενέργειες περιλαμβάνουν ουδετεροπενία, θρομβοπενία, λευκοπενία, αναιμία και λεμφοπενία. Οι πιο σπάνιοι αλλά σοβαροί κίνδυνοι που σχετίζονται με τη χρήση του beti-cel είναι η καθυστερημένη μεταμόσχευση αιμοπεταλίων που μπορεί να προκαλέσει θρομβοπενία και αιμορραγία, αποτυχία μεταμόσχευσης ουδετερόφιλων, και η ογκογένεση και αιματολογικές κακοήθειες.
Βιβλιογραφία
- American Society of Gene + Cell Therapy (ASGCT) 2022. [online] Available at <https://patienteducation.asgct.org/gene-therapy-101/gene-therapy-basics>. [Accessed: 28 March 2023].
- American Gene Technologies (AGT) 2023. Raising Awareness for Rare Disease Day. Available at: <https://www.americangene.com/blog/>. [Accessed: 28 March 2023]
- Arielle L. Langer, Erica B. Esrick; β-Thalassemia: evolving treatment options beyond transfusion and iron chelation. Hematology Am Soc Hematol Educ Program2021; 2021 (1): 600–606. doi: https://doi.org/10.1182/hematology.2021000313
- Asghar AA, Khabir Y, Hashmi MR. Zynteglo: Betibeglogene autotemcel – An innovative therapy for β- thalassemia patients. Ann Med Surg (Lond). 2022 Sep 13;82:104624. doi: 10.1016/j.amsu.2022.104624. PMID: 36268391; PMCID: PMC9577626.
- Cappellini MD., Porter J., Origa R., Forni GL., Voskaridou E., Galactéros F., Taher AT., Arlet JB., Ribeil JA., Garbowski M., Graziadei G., Brouzes C., Semeraro M., Laadem A., Miteva D., Zou J., Sung V., Zinger T., Attie KM., Hermine O. Sotatercept, a novel transforming growth factor β ligand trap, improves anemia in β-thalassemia: a phase II, open-label, dose-finding study. Haematologica. 2019 Mar;104(3):477-484. doi: 10.3324/haematol.2018.198887. Epub 2018 Oct 18. PMID: 30337358; PMCID: PMC6395345.
- Cosenza LC, Gasparello J, Romanini N, Zurlo M, Zuccato C, Gambari R, Finotti A. Efficient CRISPR-Cas9-based genome editing of β-globin gene on erythroid cells from homozygous β039-thalassemia patients. Mol Ther Methods Clin Dev. 2021 Apr 3;21:507-523. doi: 10.1016/j.omtm.2021.03.025. PMID: 33997100; PMCID: PMC8091488
- Eggimann, Beuzard, Ribeil, Bourget, Borwornpinyo, Hongeng, Hacein-Bey, Cavazzana, Leboulch, Payen, Gene Therapy of the β-Hemoglobinopathies by Lentiviral Transfer of the β(A(T87Q))-Globin Gene, Human Gene Therapy, Volume 2
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010 May 21;5:11. doi: 10.1186/1750-1172-5-11. PMID: 20492708; PMCID: PMC2893117.
- Gonçalves GAR, Paiva RMA. Gene therapy: advances, challenges and perspectives. Einstein (Sao Paulo). 2017 Jul-Sep;15(3):369-375. DOI: 10.1590/S1679-45082017RB4024. PMID: 29091160; PMCID: PMC5823056.
- HEMATOLOGY BMT INSTITUTE INTERNATIONAL (HBII) 2023. Effects of High Hemoglobin on Cancer. Available at: <https://hematologybmt.com/effects-of-high-hemoglobin-on-cancer>. [Accessed: 28 March 2023].
- Kohne E. Hemoglobinopathies: clinical manifestations, diagnosis, and treatment. Dtsch Arztebl Int. 2011 Aug;108(31-32):532-40. doi: 10.3238/arztebl.2011.0532. Epub 2011 Aug 8. PMID: 21886666; PMCID: PMC3163784.
- Langer AL, Esrick EB. β-Thalassemia: evolving treatment options beyond transfusion and iron chelation. Hematology Am Soc Hematol Educ Program. 2021 Dec 10;2021(1):600-606. doi: 10.1182/hematology.2021000313. PMID: 34889443; PMCID: PMC8791140.
- Muncie HL Jr, Campbell J. Alpha and beta thalassemia. Am Fam Physician. 2009 Aug 15;80(4):339-44. PMID: 19678601.
- Nualkaew T, Sii-Felice K, Giorgi M, McColl B, Gouzil J, Glaser A, Voon HPJ, Tee HY, Grigoriadis G, Svasti S, Fucharoen S, Hongeng S, Leboulch P, Payen E, Vadolas J. Coordinated β-globin expression and α2-globin reduction in a multiplex lentiviral gene therapy vector for β-thalassemia. Mol Ther. 2021 Sep 1;29(9):2841-2853. doi: 10.1016/j.ymthe.2021.04.037. Epub 2021 May 1. PMID: 33940155; PMCID: PMC8417505.
- Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005 Sep 15;353(11):1135-46. doi: 10.1056/NEJMra050436. PMID: 16162884.
- Somerville, MA; bluebird bio, Inc.; August 2022. 2. Negre O, Eggimann A-V, Beuzard Y, et al. Gene therapy of the β-hemoglobinopathies by lentiviral transfer of the βA-T87Q-globin gene. Human Gene Therapy. 2016;27(2):148–165. 3. Pawliuk R, Westerman KA, Fabry ME, et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. 2001;294:2368–2371
- Zakaria NA, Bahar R, Abdullah WZ, Mohamed Yusoff AA, Shamsuddin S, Abdul Wahab R, Johan MF. Genetic Manipulation Strategies for β-Thalassemia: A Review. Front Pediatr. 2022 Jun 15;10:901605. doi: 10.3389/fped.2022.901605. PMID: 35783328; PMCID: PMC9240386.
- Gene Therapy Testing Services. Available at <https://www.eurofins.com/biopharma-services/product-testing/services-by-modality/gene-therapy-testing-services/>. [Accessed: 28 March 2023].
- Δήμητρα Μπασδάνη (2020). Μία σύντομη γνωριμία με την γονιδιακή θεραπεία. Available at <https://www.truemed.gr/epistimi/mia-suntomh-gnorimia-me-ti-gonidiaki-therapeia/>. [Accessed: 28 March 2023].